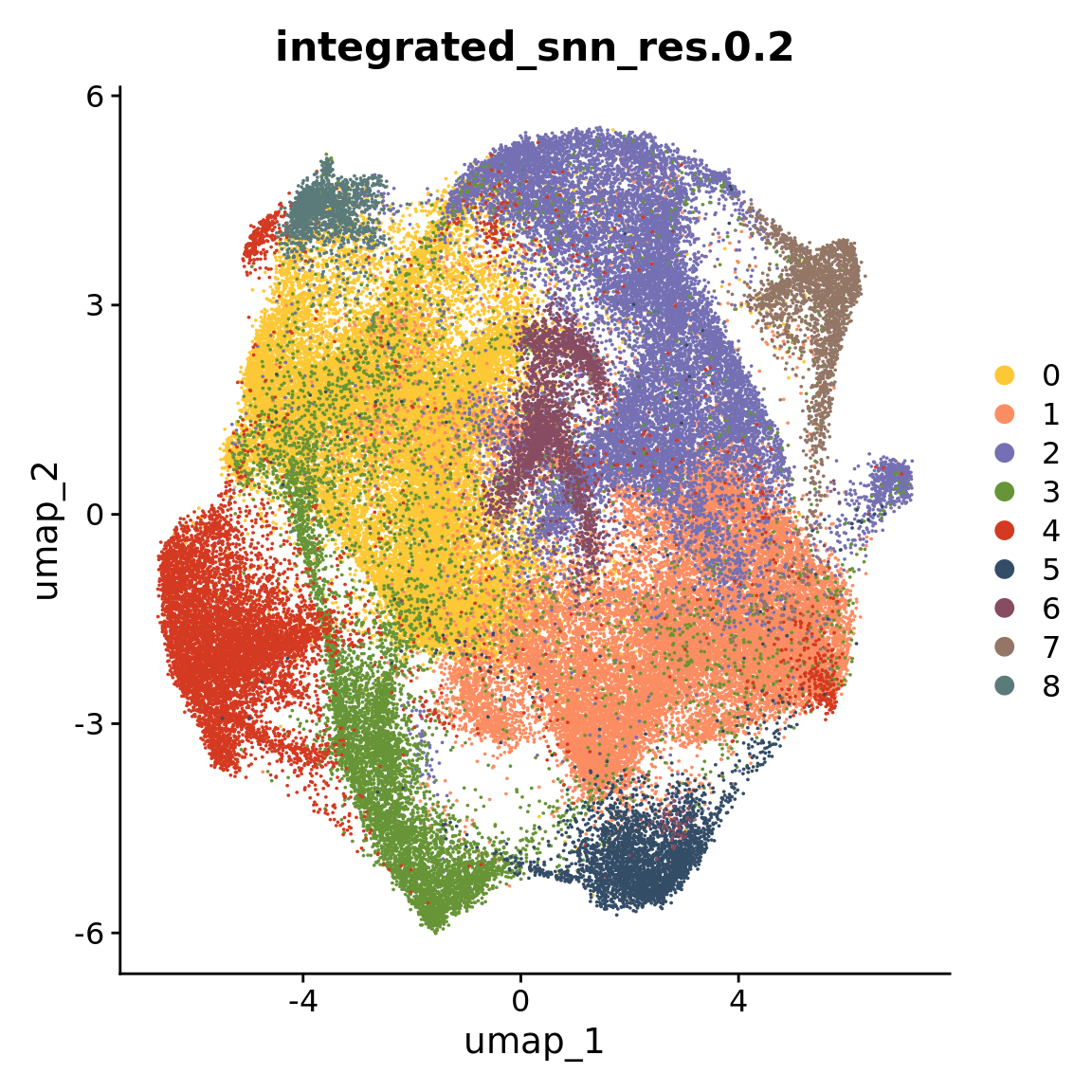
The following script depicts the identification and characterization of the nine populations within the HuMicA, and relates to the results represented in Figure 1 of the paper.
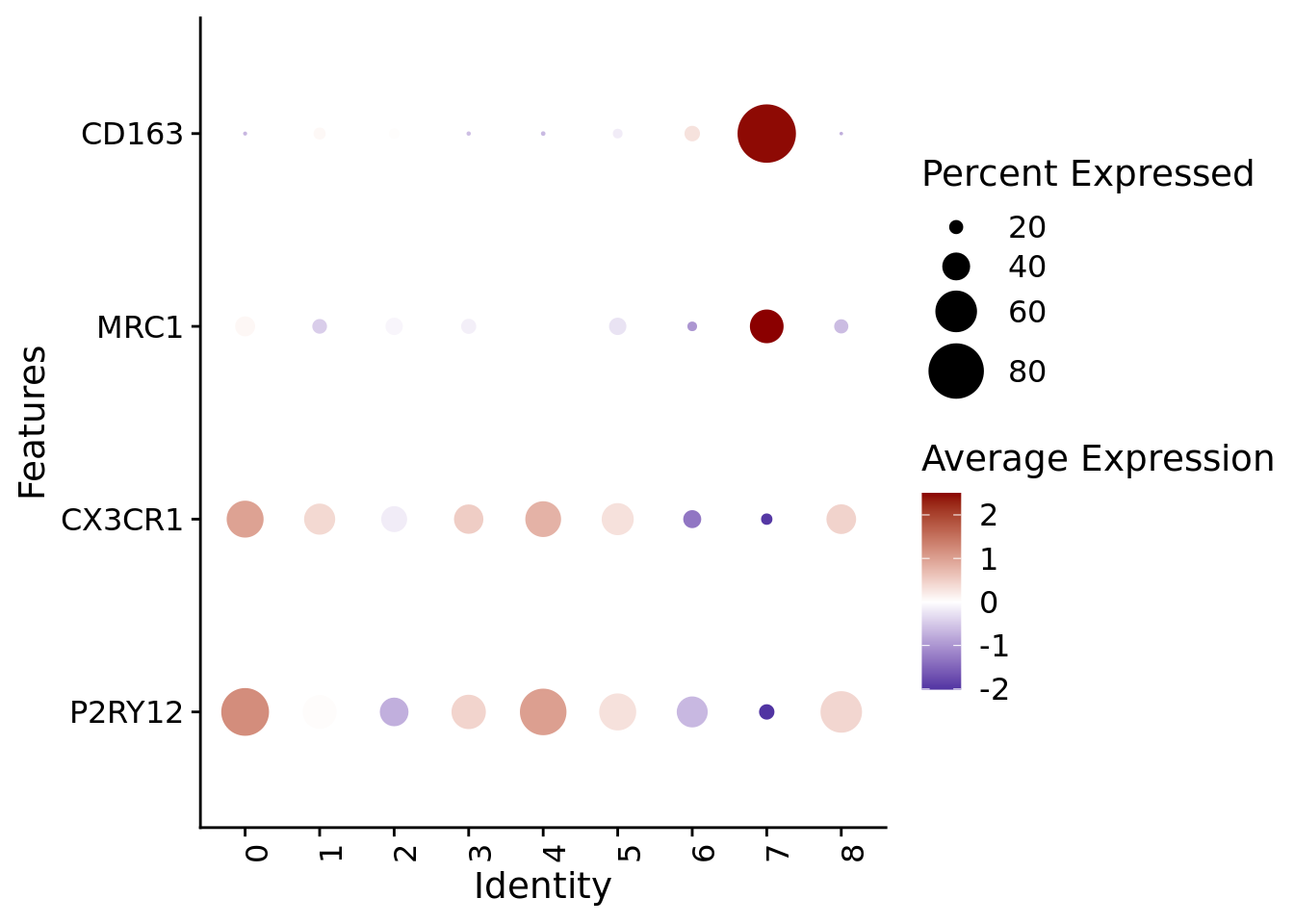
The prevalence of border-associated macrophages within the HuMicA was tested by checking the expression of canonical microglia (P2RY12, CX3CR1) and macrophage markers (MRC1, CD163). Based on this, we annotated cluster 7 as macrophages.
DefaultAssay(Humica)<-"RNA"Humica <-NormalizeData(Humica) #gene expression is evaluated using the normalized "RNA" assay# DotplotDotPlot(Humica, features =c("P2RY12","CX3CR1", "MRC1","CD163"), dot.scale =10, group.by ="integrated_snn_res.0.2") +scale_colour_gradient2(low ="darkblue", mid ="white", high ="darkred")+#scale_color_viridis_c() +theme(axis.text.x =element_text(angle=90, hjust =0))+coord_flip()
Scale for colour is already present.
Adding another scale for colour, which will replace the existing scale.
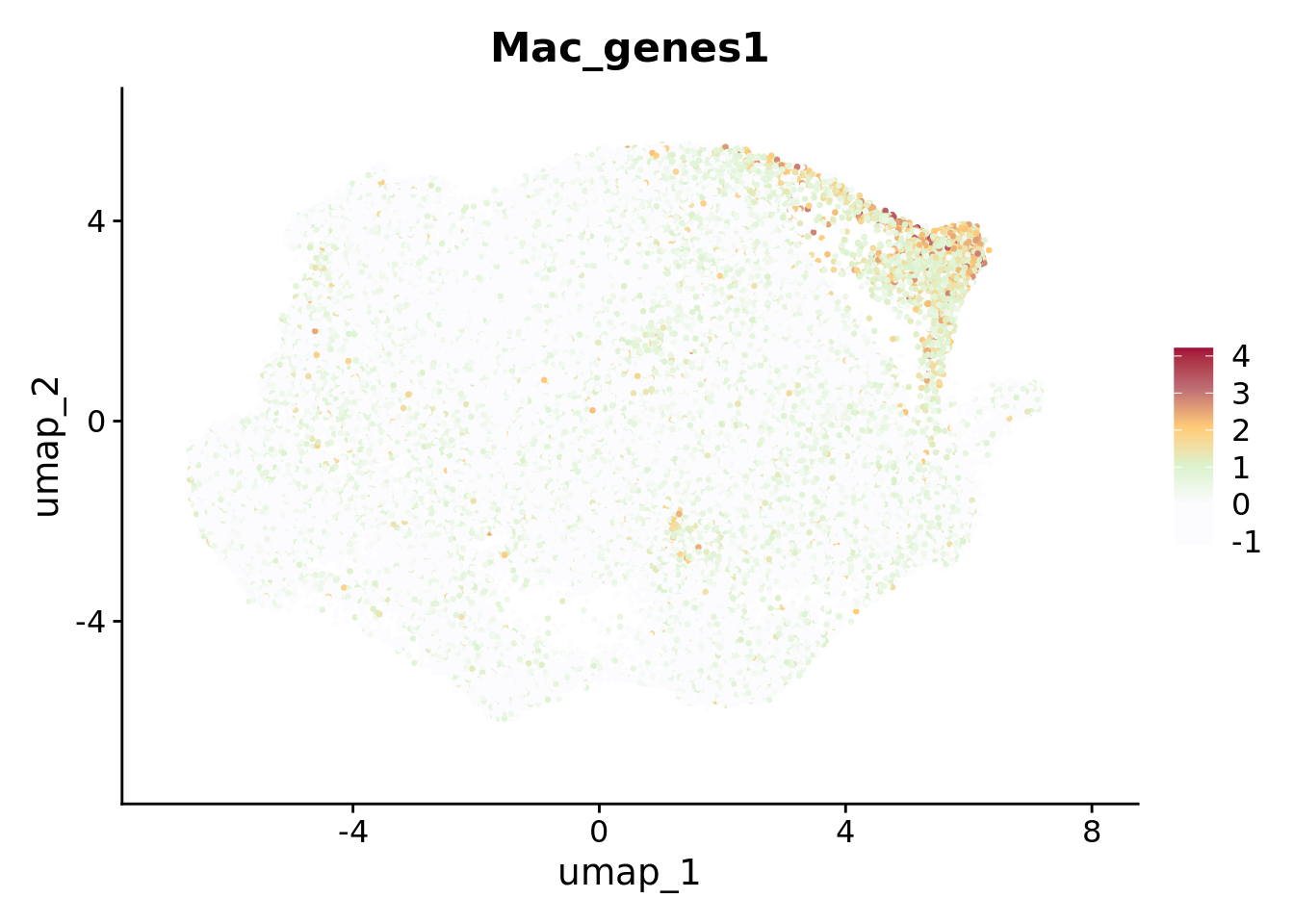
# Feature plot of the Module Score of macrophage markersMac_genes <-c("MRC1","CD163")Humica <-AddModuleScore(Humica,features =list(Mac_genes),name='Mac_genes')FeaturePlot(Humica, features ='Mac_genes1', label = F, repel =TRUE,pt.size =0.5) &scale_colour_gradientn(colours =c("#FCFCFF","#FCFCFF","#DCF2CE","#FFCB77","#BD6B73","#A30B37"))
Scale for colour is already present.
Adding another scale for colour, which will replace the existing scale.
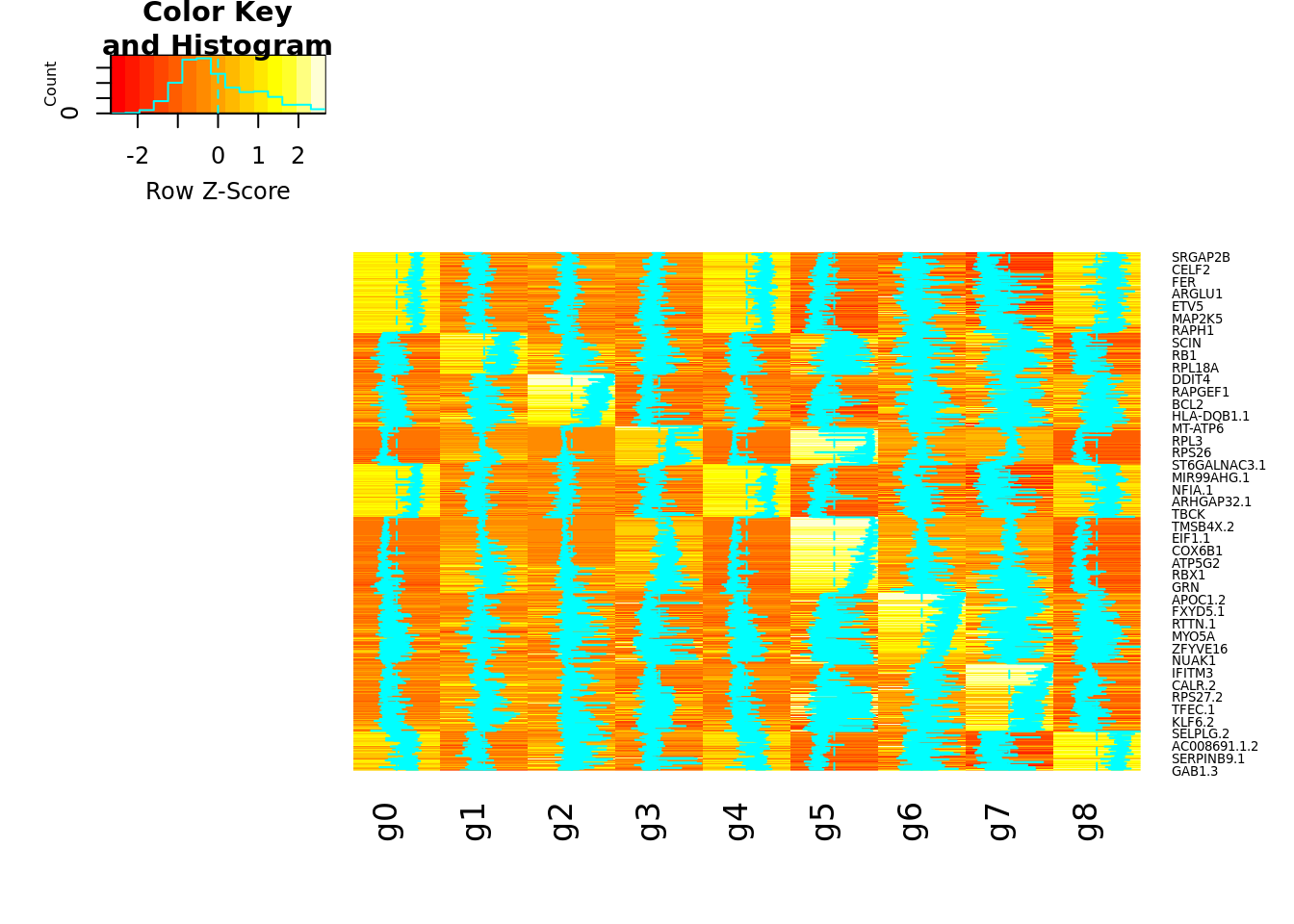
Heatmap of the cluster markers
The markers for each cluster were calculated using FindAllMarkers with the MAST test, min.pct=0.25 and logfc.theshold=0.25. Significant markers were considered for an adj p value (FDR) < 0.05.
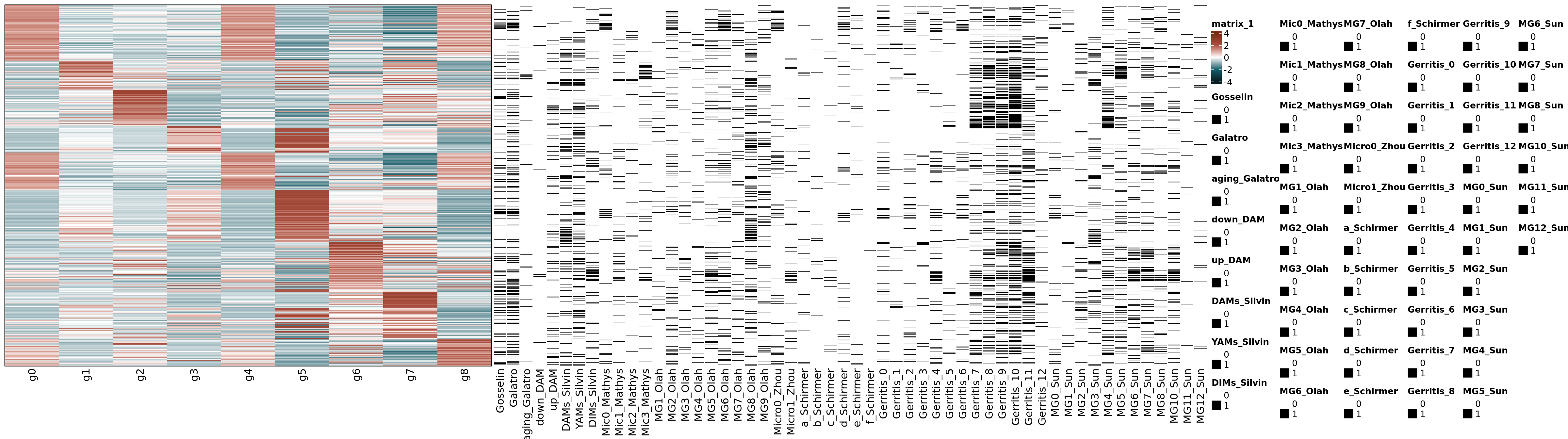
The heatmap represents the average gene expression per cluster, which consists of the average expression of all cells/nuclei annotated to each cluster. The heatmap.2 function from gplots is initially used to obtain the z-scores. Then the matrix with the z-scores is represented by the Heatmap function of ComplexHeatmap. In addition, the matching between the genes in the heatmap and microglia-related gene signatures collected from the literature is represented on the left panel. Each black lines represents the presence of one of the markers within each geneset/signature. The “Genesets_literature.xlsx” file has bee added to this repository (“Support data”).
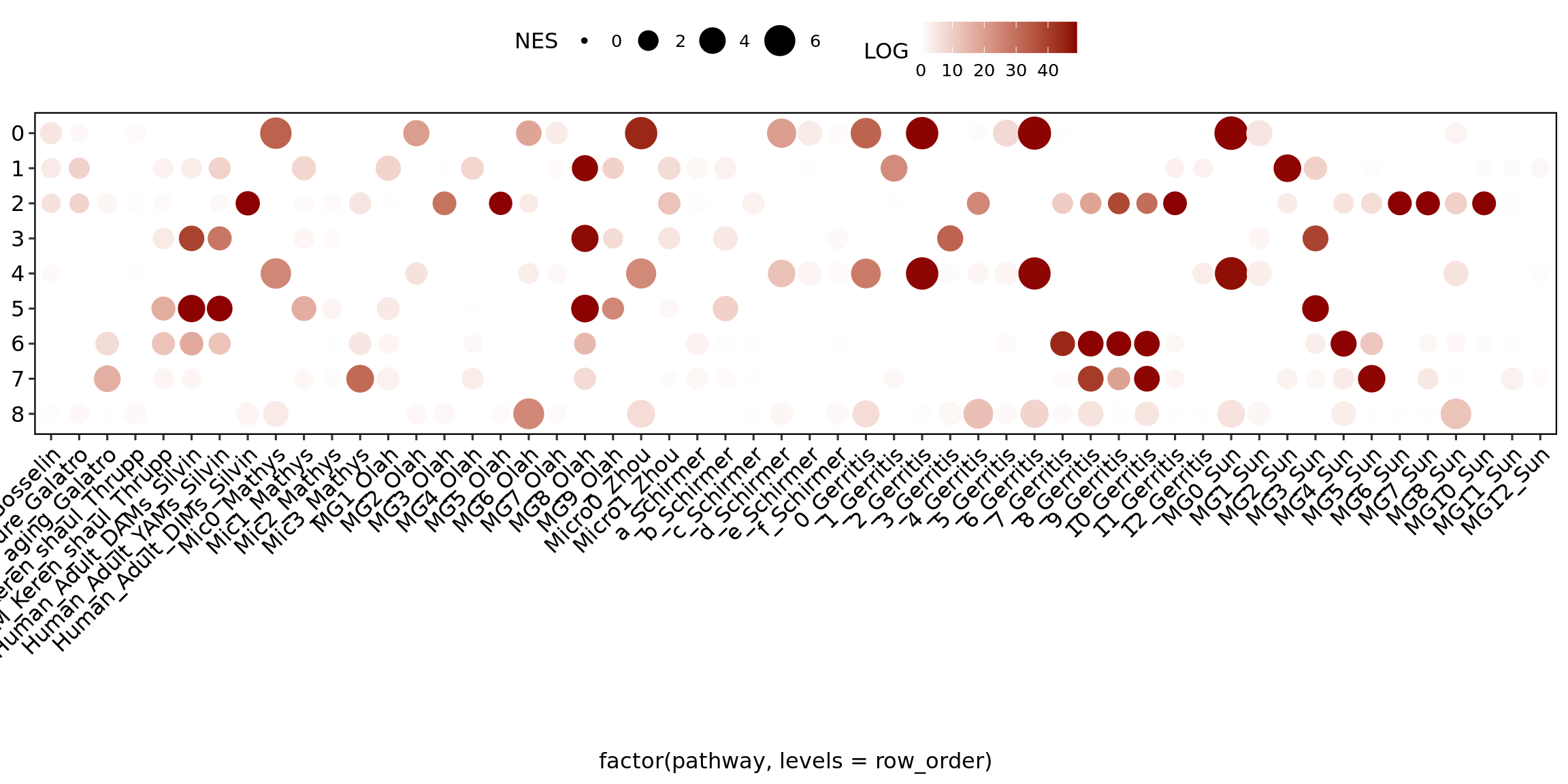
Gene set enrichment analysis (GSEA) of the signatures from the literature within each cluster
GSEA was calculated using the fgsea package. The results from FindAllMarkers with a less significant cutoff were used as cutoff. This intends to account for the broad spectrum of differential expression and not only the significant differentially expressed markers (only.pos = F, min.pct = 0.1, logfc.threshold = 0.0, test.use = “MAST”). Of note, both up and downregulated markers were considered. The avg_log2FC and the p_val_adj were used for ranking.